来源:转载风云药谈 | 发表时间:2024-10-22
10月18日,国家药监局党组书记、局长李利主持召开会议,研究部署开展生物制品分段生产改革试点工作,审议通过《生物制品分段生产试点工作方案》。
美国的法规及要求:《联邦食品、药品和化妆品法案(FD&C Act)》规定了药品生产、包装、储运等各环节的质量管理要求;《药品生产质量管理规范(cGMP)》详细规定了药品生产各环节的质量控制标准;《药品供应链安全法案(DSCSA)》要求药品从原料到终端产品的全程可追溯,确保供应链安全。
欧盟的法规及要求:《欧洲药品生产质量管理规范(EU GMP)》规定了药品生产各环节的质量管理要求,包括原料药、制剂、包装等;《欧洲药品上市许可指令(Directive 2001/83/EC)》要求药品生产必须符合GMP标准,并对生产企业资质进行审查。
其他国家的法规及要求:日本《药品、医疗器械等的质量、有效性及安全性的确保等;加拿大《食品和药品法(Food and Drugs Act)》;澳大利亚《药品生产质量管理规范(PIC/S GMP)》
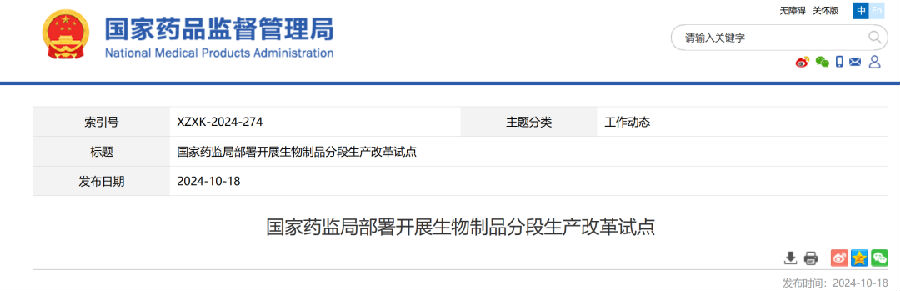
试点工作基于我国生物医药产业发展现状和监管实际,积极回应产业发展需求,以委托生产方式探索部分创新、临床急需等生物制品的分阶段生产,有利于进一步激发企业研发创新活力,促进药品研发生产专业化分工,提升创新和临床急需生物制品的供应保障能力,更好满足广大群众用药需求。
《生物制品分段生产试点工作方案》对试点范围、工作实施步骤、时间安排、监督管理要求以及保障措施等进行了部署,将于近日发布。
国家药监局将加强统筹协调,在审评审批、检查、检验、上市后监管等方面加大对试点省份支持指导力度,全力推动试点工作开展。
对生物制剂创新企业,尤其是初创企业非常利好。